Senescence Induced Inflammation (SASP) May Promote Cell and Telomere Damage, Leading to Allodynia.
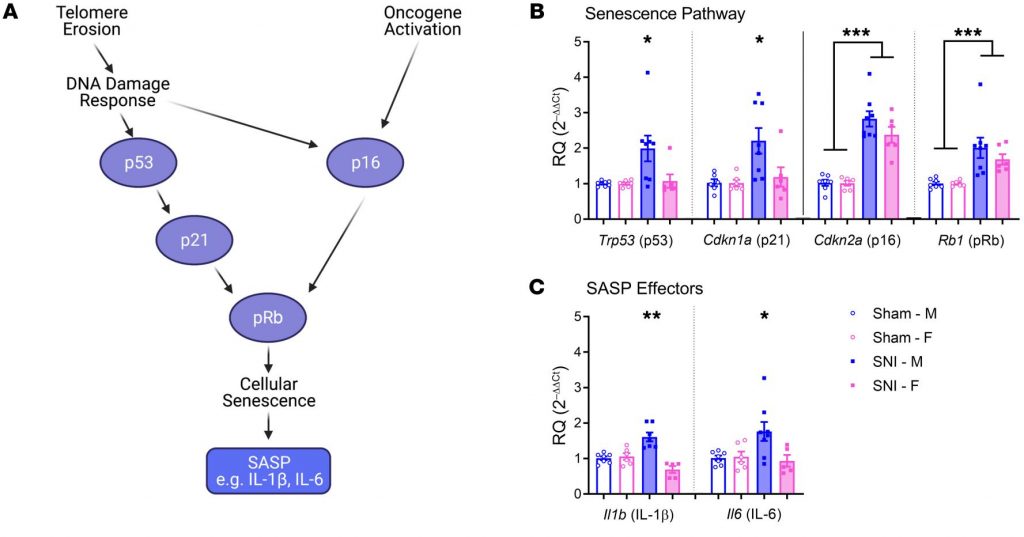
“Expression of senescence pathway and SASP effector genes in the spinal cords of mice of both sexes 6 months after sham or SNI surgery. “
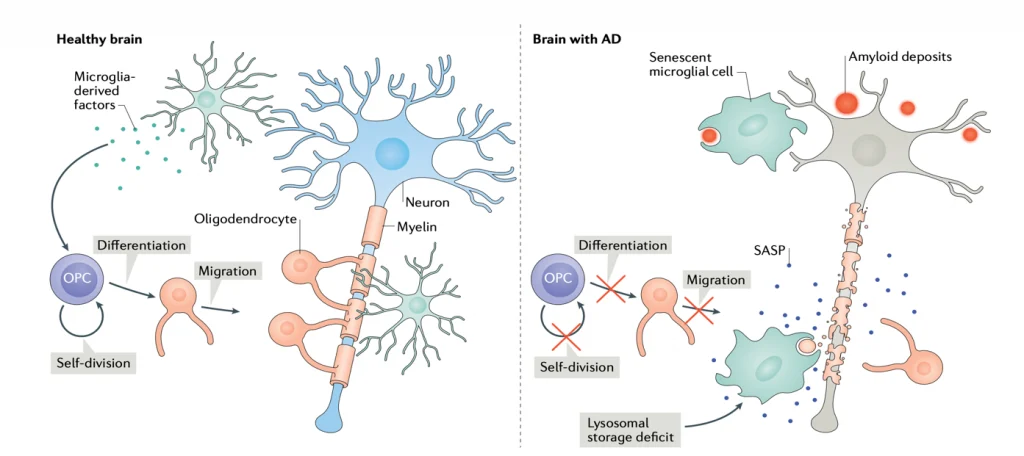
(SASP is inflammation secreted by senescent cells.)
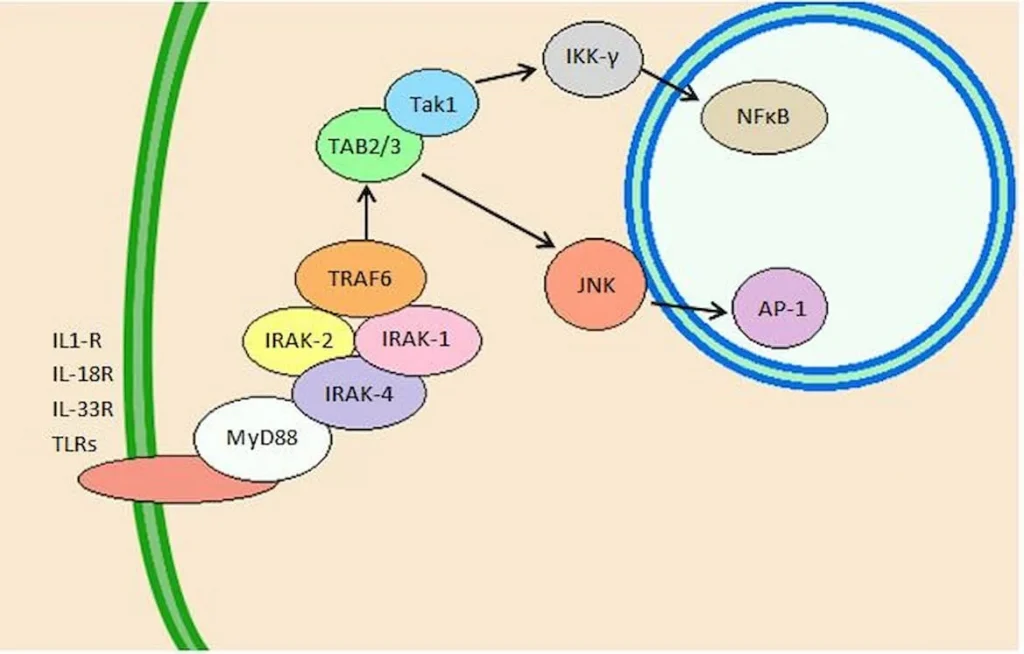
“Peripheral nerve injury produces cellular senescence in the spinal cord of mice at time points long after injury. Reduced TL can result in a persistent DNA damage response leading to cellular senescence — a state of cell cycle arrest/withdrawal, deregulated cellular metabolism, and macromolecular damage — and senescent cells in turn release a diverse set of cytokines, growth factors, proteases, and extracellular matrix components, together known as the senescence-associated secretory phenotype (SASP) or senescent-messaging secretome. Many of these SASP-related compounds are proinflammatory, and well known to produce or facilitate pain, especially when released in the spinal cord. We gave new cohorts of young male and female mice SNI or sham surgeries and harvested lumbar spinal cord tissues from these animals 12–14 months later, or 2 months later.” (1)